-
Linearized orbital-free embedding potential in self-consistent calculations
M. Dulak, J.W. Kaminski and T.A. Wesolowski
International Journal of Quantum Chemistry, 109 (9) (2009), p1886-1897


DOI:10.1002/qua.22011 | unige:3175 | Abstract | Article HTML | Article PDF
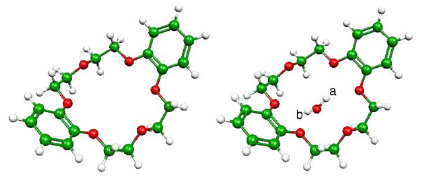
Conventionally, solving one-electron equations for embedded orbitals[Eqs. (20) and (21) in Wesolowski and Warshel, J Phys Chem, 1993, 97, 8050] proceedsby a self-consistent procedure in which the whole effective potential, including itsembedding component, is updated in each iteration. We propose an alternative scheme(splitSCF), which uses the linearized embedding potential in the inner iterative loop andthe outer-loop is used to account for its deviations from linearity. The convergence ofthe proposed scheme is investigated for a set of weakly bound intermolecularcomplexes representing typical interactions with the environment. The outer loop isshown to converge very fast. No more than 3-4 iterations are needed. Errors due toskipping the outer loop completely and using the electron density obtained in theabsence of the environment in the linearized embedding potential are investigated indetail. It is shown that this computationally attractive simplification, used already innumerical simulations by others, is adequate not only for van der Waals and hydrogen-bondedcomplexes but even if the complex comprises charged components, i.e., wherestrong electronic polarization takes place. In charge-transfer type of complexes, largerchanges of electron of density upon complex formation occur and the abovesimplification is not recommended. Figure (a) The splitSCF scheme: In the inner loop(i-index), the embedding potential vemb[ÏA,ÏB]is evaluated for A taken from the previous iteration in theouter loop (j-index) and remains constant, whereas thevKS[ÏA] component is recalculated as A changes. (b) The conventional SCF scheme: Both vKS[ÏA] andvemb[ÏA,ÏB] are recalculated as A changes.